Experimental Features
The Experimental section of the web app provides tools for quick, one-off tasks — useful for testing new features.
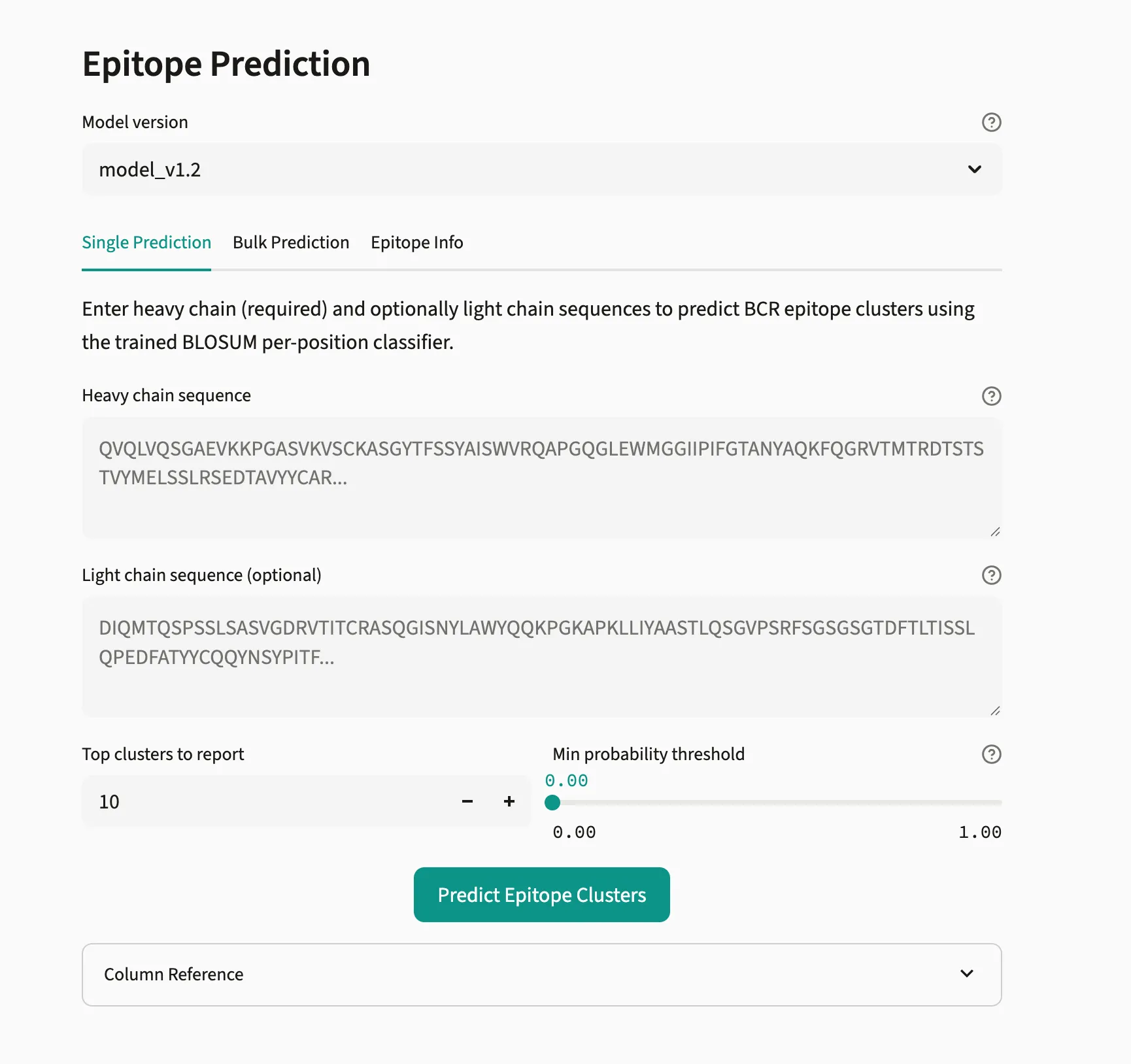
Single Epitope Prediction
Section titled “Single Epitope Prediction”Predicts BCR epitope clusters for a single antibody using a trained classifier.
- Select a Model version from the dropdown (current:
model_v1.2) - Enter the Heavy chain sequence (required) — full amino acid sequence
- Optionally enter a Light chain sequence to improve prediction accuracy
- Set Top clusters to report (default: 10)
- Set Min probability threshold (recommended: 0.5)
- Click Predict Epitope Clusters
Result columns
Section titled “Result columns”| Column | Description |
|---|---|
rank | Rank for this query (1 = highest probability) |
mean_prob | Mean classifier probability across all reference antibodies in this cluster |
msa_cluster_id | Fine-grained epitope cluster ID (MSA-based) |
epitope_cluster | Broader epitope cluster grouping |
ag_cluster_id | Antigen cluster ID |
antigen | Antigen name associated with the predicted cluster |
n_ref_abs | Number of reference antibodies in this cluster |
rep_ids | Top-3 representative reference antibody IDs |
Single Sequence Search
Section titled “Single Sequence Search”Search a single amino acid sequence against one or more reference databases using nearest-neighbor retrieval. Chain type and species are auto-detected from the input sequence.
- Enter an amino acid Query sequence — chain type and species are detected automatically
- Select one or more Databases to search (default: SAbDab). Once the chain type is known, the list is filtered to the matching receptor class — BCR databases for
IGH/IGK/IGLand TCR databases forTRA/TRB/TRD/TRG. All databases stay available until a chain type is determined. - Set Nearest neighbors to retrieve — candidates fetched from the indexes (default: 1000)
- Set Results to keep — top hits returned after scoring (default: 100)
- Choose Search mode: Full paratope (default) or CDR3 only
- Click Search
Results can be downloaded as CSV.
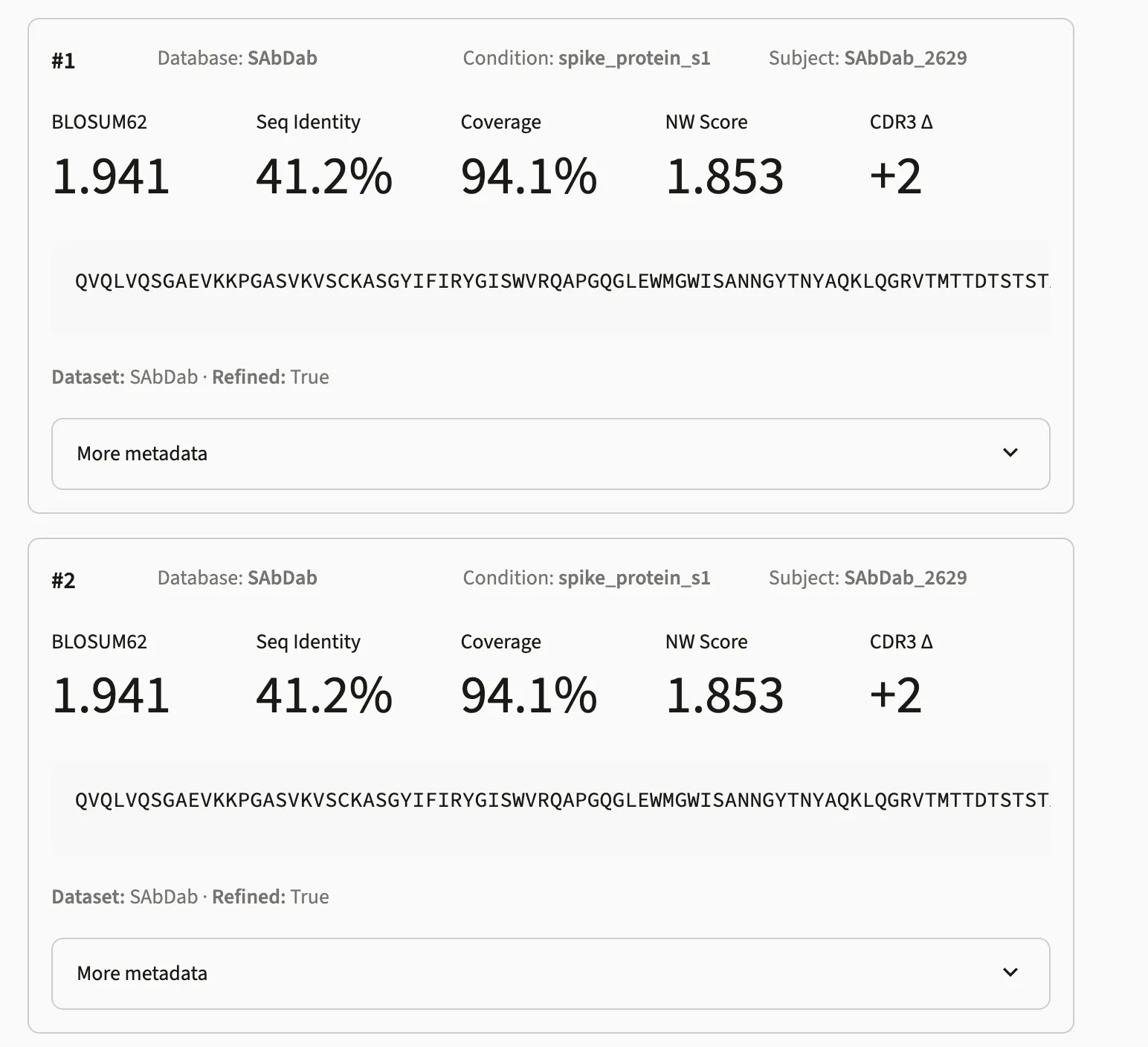
Result columns
Section titled “Result columns”| Column | Description |
|---|---|
database | Source database for this match |
aligned_sequence | Pseudo-sequence alignment of the matched target |
distance | Raw levenshtein distance — lower is more similar |
sequence_identity | Fraction of matching residues at non-gap positions (0–1) |
coverage | Fraction of positions where both sequences have a residue (0–1) |
blosum62_score | Mean per-position BLOSUM62 score — higher is more similar |
nw_score | Needleman–Wunsch score normalised by the longer sequence — higher is more similar |
cdr3_length_diff | CDR3 length difference (query − target; 0 = equal length) |
deamidation | Count of deamidation motifs (NG, NS) in the padded CDR regions |
isomerization | Count of isomerization motifs (DG, DS) in the padded CDR regions |
oxidation | Count of oxidation-prone residues (M, W) in the padded CDR regions |
refined | Whether CDR3 gap positions were repositioned via BLOSUM62 sliding-window refinement |
glycosylation | Potential N-linked glycosylation sequons (N-X-S/T, X ≠ Pro) scanned across the full target sequence — count and 1-based positions |
dataset_name | Source dataset the target sequence belongs to |
condition | Condition label associated with the target sequence |
subject_idx | Subject identifier within the source dataset |
| extra columns | Database-specific metadata (e.g. antigen.epitope, v.alpha) — fields listed under Database Info |
When to use vs. full pipelines
Section titled “When to use vs. full pipelines”| Scenario | Use |
|---|---|
| Testing one sequence | Experimental tools |
| Analyzing a full cohort (100+ sequences) | Bulk pipeline jobs |
| Need results saved on cloud | Bulk pipeline jobs |
| Quick sanity check before a batch run | Experimental tools |